High-throughput computational screening of MOFs for gas adsorption and separation
By Tang Hongjian, Department of Chemical & Biomolecular Engineering, Faculty of Engineering
Abstract
Metal–organic frameworks (MOFs) have emerged as a versatile material due to their structural diversity and facile tunability. For practical application, it is highly desired to identify the best candidates with targeted properties among ~100,000 MOFs that have been experimentally reported. High-throughput computational screening paves the way to such demand, which has been successfully implemented in a broader research domain. Such a method has been validated as time-efficient and technically practical. It shortlists candidate structures from the whole MOF database to boost experimental synthesis and performance evaluation. Our group supervisor Prof. Jiang Jianwen is specialized in this field.
1. Introduction
Metal-organic frameworks (MOFs) are a relatively new class of porous, crystalline materials with a broad range of applications. MOFs have substantially more diversity and multiplicity than any other types of porous materials (e.g., zeolites). Their crystalline structures, surface areas, pore sizes, and shapes can be rationally tuned. As a consequence, they have attracted extensive interests for many potential applications in separation, storage, catalysis, etc. Owing to their facile tunability ~100,000 MOFs have been experimentally synthesized heretofore, displaying diverse pore geometries, surface chemistries and, corresponding practical properties. For the application of gas separation, it is highly desired to identify the best candidates with optimal separation properties for a given gas mixture among the ~100,000 experimentally reported MOFs. Nevertheless, experimentally testing of the enormous number of MOFs is technically formidable and economically infeasible. In the past several years, computational methods were increasingly employed to screen MOFs. Emerging studies of high-throughput computational screening of MOF database were reported for the applications of CO2 capture, CH4 and H2 storage, hydrocarbons separation, toxic gas removal, etc. As such methods have been validated as time-efficient and technically practical to shortlist candidate structures from the whole MOF database for further experimental synthesis and performance evaluation.
2. MOF database
Database containing reliable MOF structures is an essential prerequisite to throughput computational screening of desired MOFs. Therefore, a handful of MOF databases have been constructed based on experimental or hypothetical materials, for instance, the Cambridge Structural Database (CSD) MOF dataset; computation-ready, experimental (CoRE) MOF database; and hypothetical MOF databases.
CSD MOF dataset collects structures determined by X-ray diffraction. Experimentally reported MOFs are deposited to CSD along with the publication of related studies. Up to now, 101445 MOFs exist in CSD MOF dataset (2020.1 CSD release version). However, MOFs collected in CSD dataset are not computation-ready. Some structural issues should be noticed or approached with caution. For instance: (1) Both free and bonded solvents exist in the pores of CSD MOFs; (2) Structural disorders like missing of hydrogen atoms. Python API scripts developed by David Fairen-Jimenez’s group enable CSD users to process the removal of solvents and exclusion of disordered MOFs throughout the dataset.
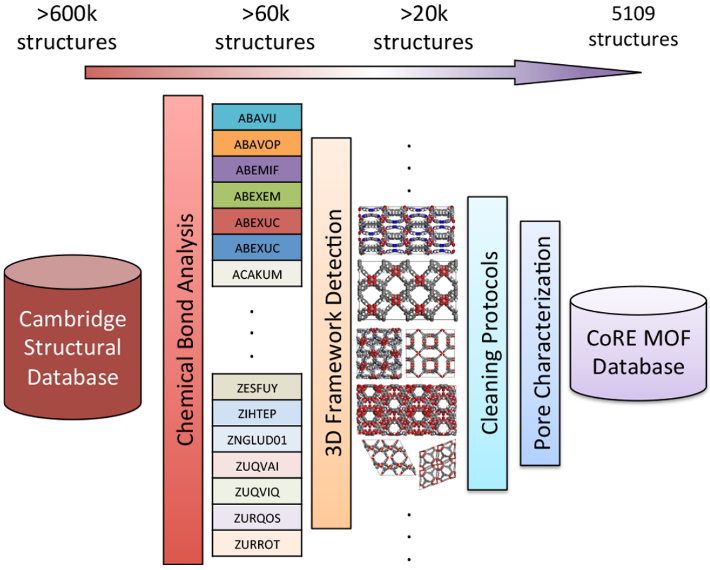
Figure 1. Schematic illustration of the CoRE MOF database construction.
CoRE-MOF database contains computation-ready MOFs, first released in 2014. As shown in Figure 1, CoRE MOFs are originally derived from CSD MOFs. Free and bonded solvents are identified and cleaned. Problematic framework structures are excluded or amended, resulting in 4764 computation-ready MOFs in version 2014. Besides, an extension of atomic point charge assignment is done for 2932 experimentally synthesized structures by using DFT-derived DDEC charge. Moreover, 879 structures in version 2014 are structurally DFT-minimized. In the updated CoRE-MOF database (version 2019), 10143 MOF structures without disorders and solvents are publicly accessible. Geometric properties like pore size, surface area, porosity are supplemented to support high-throughput computational studies.
Hypothetical MOFs (hMOFs) are constructed to mimic the real-world assembly of secondary building units (metal clusters and organic linker) following different coordination manners. Therefore, hypothetical MOF databases contain more enormous MOF quantities than experimentally reported ones. hMOFs containing 137953 structures were reported by Randall Snurr’s group; while 325000 hMOFs were generated by Berend Simit’s group. These hMOFs databases have been successfully applied for high-throughput computational studies and demonstrated to be reliable by experimental conformations.
3. High-throughput computational screening methods
Typically, high-throughput computational screening of MOF database is based on geometrical analysis and molecular simulations. The geometrical properties like pore size, surface area, porosity could be facilely predicted by Zeo++, Poreblazer, etc. For molecular simulations, interactions between MOF framework and gas molecules in the adsorption phase are mathematically described by a potential energy model based on empirical or quantum-derived force fields. Molecular simulations of adsorption in MOFs is a result of time-efficiency and good accuracy if the force field can fit well to experimental properties or ab initio-derived energies. A typical Lennard-Jones potential is described as following:
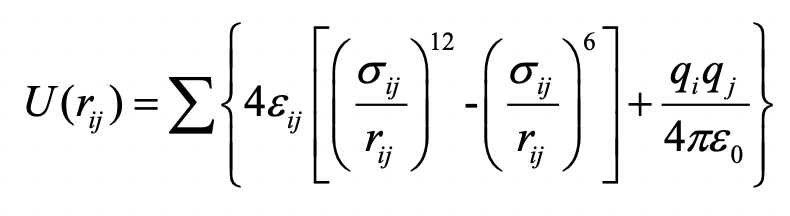 Based on LJ potential, Universal and, DREIDING force fields are widely applied for MOF atoms; while TraPPE force fields are specified for a wide range of gas molecules. Generally, employing rational ensembles, Monte Carlo (MC) simulations and molecular dynamics (MD) are specialized in predicting adsorption-based and diffusion-based properties in MOFs, respectively.
Based on LJ potential, Universal and, DREIDING force fields are widely applied for MOF atoms; while TraPPE force fields are specified for a wide range of gas molecules. Generally, employing rational ensembles, Monte Carlo (MC) simulations and molecular dynamics (MD) are specialized in predicting adsorption-based and diffusion-based properties in MOFs, respectively.
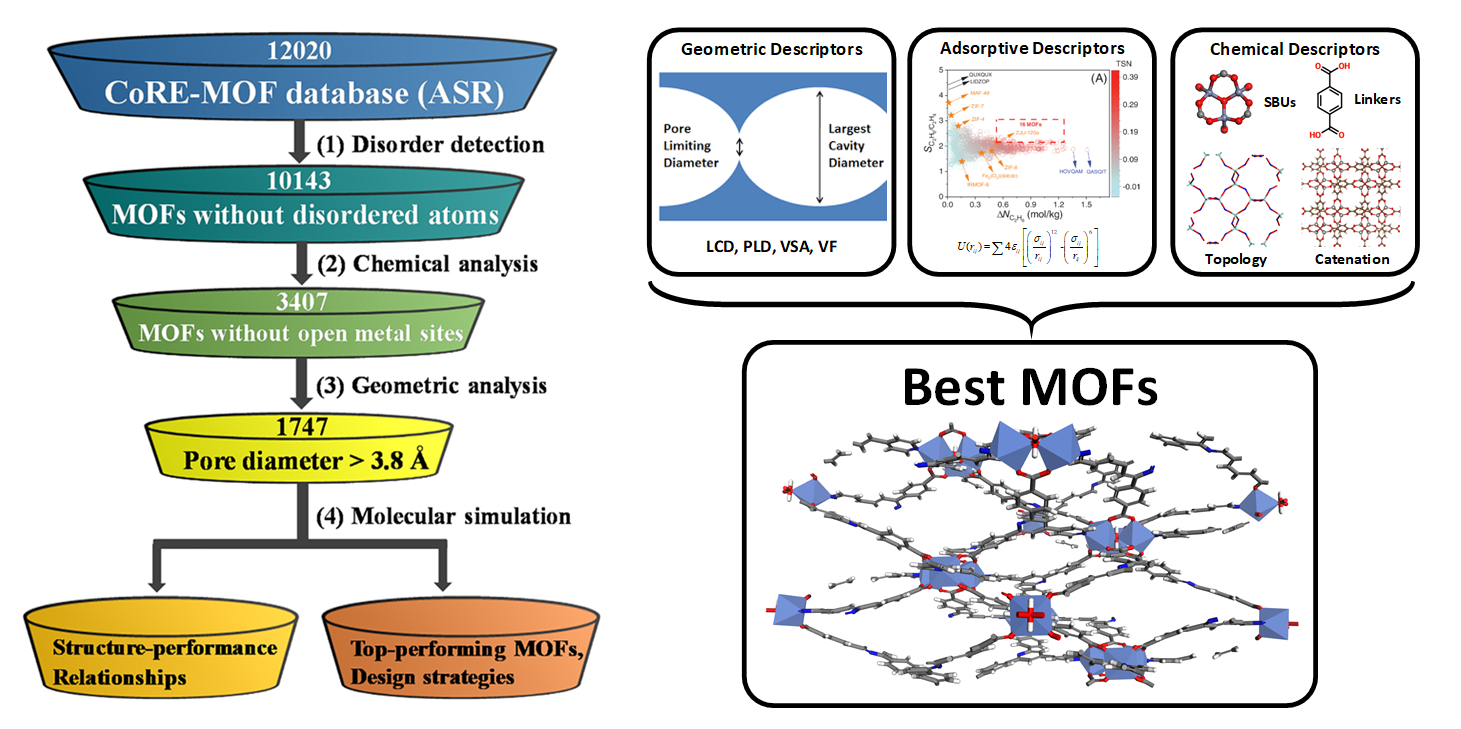 Figure 2. Left: Screening workflow to identify top-performing MOFs. Right: Geometrical, adsorptive and, chemical descriptors to target MOF candidates.
Figure 2. Left: Screening workflow to identify top-performing MOFs. Right: Geometrical, adsorptive and, chemical descriptors to target MOF candidates.
Figure 2 shows a typical case of high-throughput computational study for C2H6-selective CoRE MOFs (doi: 10.1002/aic.17025) by us via the integration of the geometric, adsorptive, and chemical descriptors. CoRE-MOF database 2019, is initially narrowed down from 12020 to 1747 MOFs via chemical and geometric analysis. Then, molecular simulations are conducted to predict adsorptive properties of C2H6/C2H4 for the remaining MOFs. Furthermore, structure–performance relationships are correlated between MOF descriptors and separation metrics, resulting in 16 top‐performing MOFs with promising C2H6-selective properties. Finally, based on identification and analysis of MOF building units (see Figure 3), 6 design strategies are proposed for the experimental synthesis of new MOFs to boost C2H6/C2H4 separation.
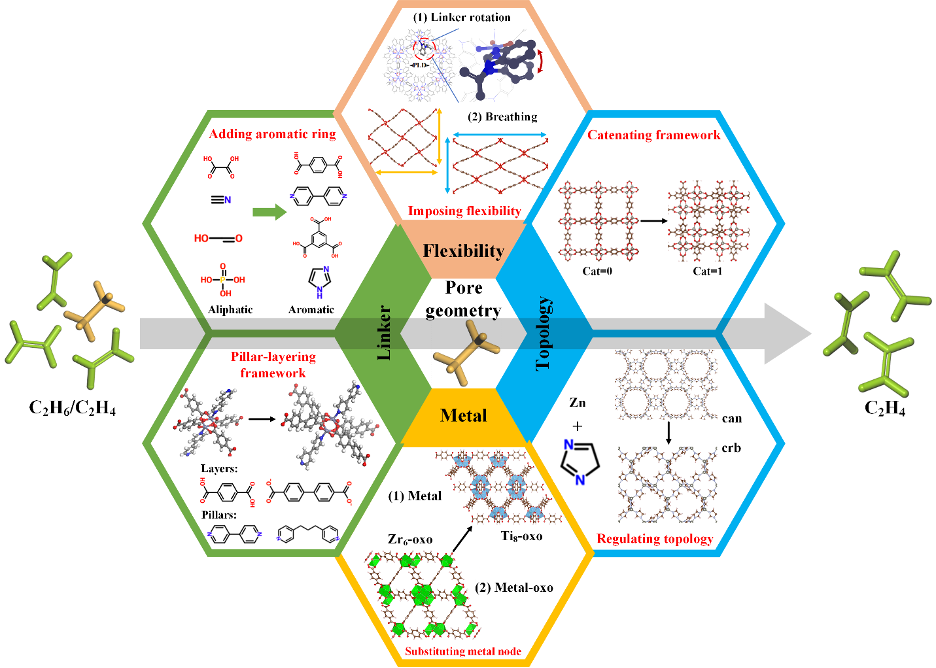 Figure 3. Six design strategies for boosting C2 separation.
Figure 3. Six design strategies for boosting C2 separation.
4. Summary
High-throughput computational screening is a brute-force strategy to identify optimal MOF structure. Whereas, it should be really practical to shortlist MOF candidates and to comprehend structure-property relationships from a molecular insight, which is technically formidable by experimental approaches.
The efforts driven by machine-learning methods would render high-throughput computational studies more efficient and reliable. More specific details of our research regarding high-throughput computational screening of MOFs can be found at Prof. Jiang Jianwen’s group website http://cheed.nus.edu.sg/stf/chejj/index.html.
Acknowledgement
I would like to thank Dr. Wang Junhong from HPC@NUS for his invitation to share my research. All my simulation tasks are conducted on the basis of HPC computational platform. HPC@NUS system offers a smooth and user-friendly experience.
I would also like express my gratitude to all staff that work for HPC, especially Mr. Jitesh Mathpal, who has supported me with code compilation and job running issues via A.S.K. A.S.K. is highly recommended for HPC beginners, which is a reliable channel to feedback issues and improve the efficiency of using HPC@NUS.
